Аномалия арнольда киари
Содержание:
Симптоматика
Синдром Арнольда-Киари I типа может длительное время протекать бессимптомно.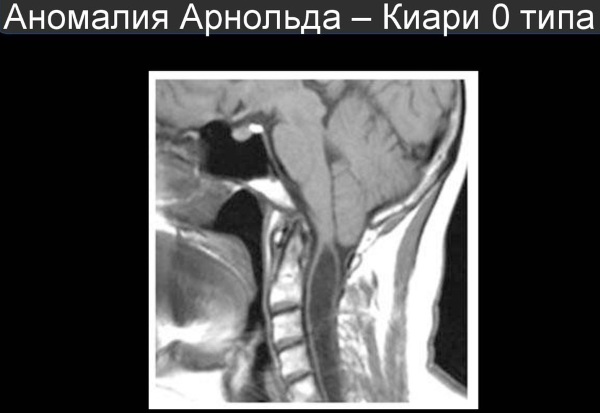
Это нарушение имеет следующие признаки:
- болезнь проявляется у подростков либо в 30-40-летнем возрасте;
- нарушения чувствительности, чаще всего в руках (40-76% пациентов);
- головная боль (наиболее распространенный симптом, отмечающийся у 47-73% больных);
- боль в шее и затылке, что усиливается из-за кашля, чихания, напряжения брюшных мышц;
- частые обмороки, падения, связанные с нарушением кровообращения спинного мозга, что вызывает резкое снижение мышечного тонуса; онемение конечностей;
- частое прекращение дыхания во сне более, чем на 10 секунд (50-70% пациентов), острая дыхательная недостаточность;
- общая слабость и снижение тонуса в конечностях;
- измененное вынуждено положение головы;
- скандирование речи (разделение на отдельные слова);
- колебательные движения глаз, преимущественно направленные вниз;
- нарушенная координация, походка, двигательная активность; рассогласованность, избыточность или недостаточность амплитуды движений;
- искривление позвоночника (50-75% пациентов);
- задержка роста и умственного развития у детей, иногда преждевременное половое созревание;
- приступообразный кашель (10-14% больных);
- нарушение мелкой моторики, дрожание конечностей;
- ухудшение слуха (одностороннее или двустороннее);
- врожденные анатомические особенности – короткая, «бычья» шея, воронкообразная грудь, асимметричная форма черепа, низкая граница оволосения на шее, деформация стоп, аномалии сосков, высокое стояние лопатки;
- раздвоение в глазах, разные размеры зрачков, слепые участки в поле зрения, опущение века;
- нарушение глотания, атрофия мышц языка, частое икание;
- редко – замедление сердечного ритма, острая сердечно-сосудистая недостаточность, гипогликемия.
Зависимо от степени поражения симптомы могут варьироваться от незначительных до ярко выраженных.
При мальформации II типа отмечаются следующие признаки:
- появление симптомов уже в период новорожденности;
- спинномозговая грыжа;
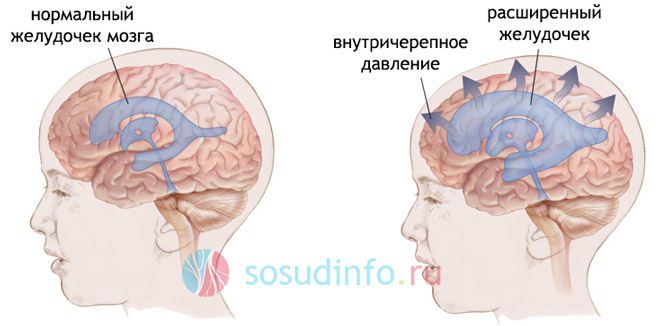
- гидроцефалия (водянка головного мозга, увеличенная окружность головы, выбухающий родничок у новорожденных, косоглазие, высокая возбудимость, рвота, вялость, головные боли и головокружения);
- приступы удушья и остановки дыхания, в том числе во сне, свистящее шумное дыхание;
- ухудшенный глотательный рефлекс, затруднения при глотании (70% пациентов);
- слабость рук и мышц лица, снижение двигательной активности;
- судорожная поза с выгибанием спины;
- потеря голоса в результате пареза голосовых связок;
- дрожание глазных яблок, направленное вниз.
Для формы III варианта характерны такие симптомы, как:
- гидроцефалия;
- сердечно-сосудистые нарушения;
- патологии желудочно-кишечного тракта, мочевыводящей системы;
- головокружение;
- неустойчивость при передвижении;
- звон в ушах;
- головная боль;
- повышенный тонус в шейных мышцах.
Лечение
При бессимптомном течении показано постоянное наблюдение с регулярным ультразвуковым и рентгенографическим исследованием. Если единственный признак аномалии – незначительные боли, пациенту назначают консервативное лечение. Оно включает разнообразные варианты с использованием нестероидных противовоспалительных средств и миорелаксантов. К наиболее распространенным НПВС относятся Ибупрофен и Диклофенак.
Нельзя самостоятельно назначать себе обезболивающие препараты, так как они имеют ряд противопоказаний (например, язвенная болезнь). При наличии какого-либо противопоказания врач подберет альтернативный вариант лечения. Время от времени назначают дегидратационную терапию. Если в течение двух-трех месяцев эффекта от такого лечения нет, проводят операцию (расширение затылочного отверстия, удаление дужки позвонка и т. д.). В этом случае требуется строго индивидуальный подход, позволяющий избежать как ненужного вмешательства, так и проволочки с операцией.
 Тактика лечения в отношении каждого пациента требует индивидуального подхода
Тактика лечения в отношении каждого пациента требует индивидуального подхода
У некоторых пациентов хирургическая ревизия является способом постановки конечного диагноза. Цель вмешательства – ликвидация сдавливания нервных структур и нормализация ликвородинамики. Такое лечение приводит к существенному улучшению у двух-трех пациентов. Расширение черепной ямки способствует исчезновению головных болей, восстановлению осязаемости и подвижности.
Симптомы
Клиника аномалии Киари зависит от ее разновидности и тяжести повреждений. Все симптомы делятся на мозжечковые (церебральные), гипертензионно-гидроцефальные, бульбарно-пирамидные, корешковые, сирингомиелитические. У ряда пациентов могут наблюдаться также признаки вертебробазилярной недостаточности.
Мозжечковый синдром включает нарушение координации движений, неконтролируемые движения глазных яблок (нистагм), ухудшение мелкой моторики пальцев, шаткость походки. К церебральным проявлениям относятся также тремор рук, расстройство речевой функции, двоение в глазах. У детей может отмечаться интеллектуальная недоразвитость.
Гипертензионно-гидроцефальные признаки обусловлены нарушением оттока спинномозговой жидкости, повышением внутричерепного давления и гидроцефалией. К ним причисляют головную боль распирающего характера, тошноту и рвоту вне зависимости от приема пищи, спазмы затылочных мышц. Пациенты нередко жалуются на плохое самочувствие по утрам, сразу после пробуждения, дискомфорт при поворотах и наклонах головы.
Бульбарно-пирамидный синдром проявляется следующими симптомами:
- заметным ослаблением мышц в руке или туловище, вплоть до паралича;
- болевым синдромом и неприятными ощущениями в затылке, причем боль усиливается при движениях головы, включая смех и чихание;
- онемением отдельных зон кожи;
- затрудненным дыханием и глотанием, нечленораздельной речью.
Симптомы вертебробазилярной недостаточности возникают из-за сдавления сосудов, что вызывает нарушение кровоснабжения и питания головного мозга. Начинаются сильные головокружения, переходящие иногда в обмороки, снижается сила в мышцах, острота зрения и слуха.
Речь идет о корешковом синдроме, если компрессии подвергаются подъязычный, блуждающий и другие нервы, проходящие в области затылка. Он проявляется онемением языка, лица, затрудненностью глотания, снижением речевой и слуховой функции.
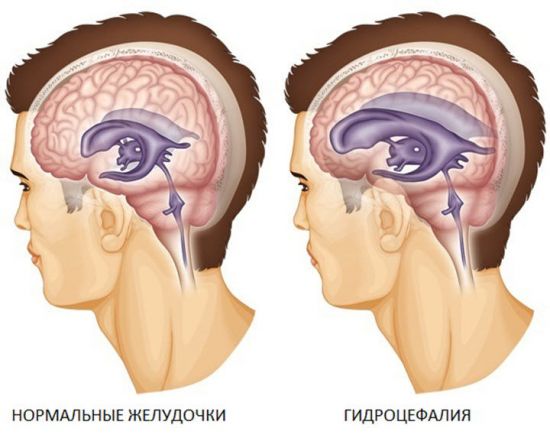 Одним из провокаторов характерных симптомов дефекта выступает гидроцефалия – чрезмерное скопление спинномозговой жидкости в желудочках головного мозга
Одним из провокаторов характерных симптомов дефекта выступает гидроцефалия – чрезмерное скопление спинномозговой жидкости в желудочках головного мозга
Сирингомиелитический синдром обуславливает возникновение и рост кисты в веществе продолговатого либо спинного мозга. Он объединяет следующие признаки: расстройство чувствительности в руках и верхней части туловища – тактильная восприимчивость может снижаться в ответ на температурные и болевые раздражители. Человек не чувствует
- боли при ожогах, ранениях и травмах;
- слабость и постепенную атрофию мышц рук, корпуса и шеи;
- деформацию суставов, искривление позвоночного столба;
- нарушение работы выделительных органов с недержанием мочи и фекалий.
Аномалия Арнольда Киари 1-4 степени может иметь разный характер течения – от абсолютно бессимптомного до бурного и молниеносного прогрессирования. Симптоматика может появиться как сразу после рождения ребенка, так и отсутствовать в течение всей жизни.
Диагностические мероприятия
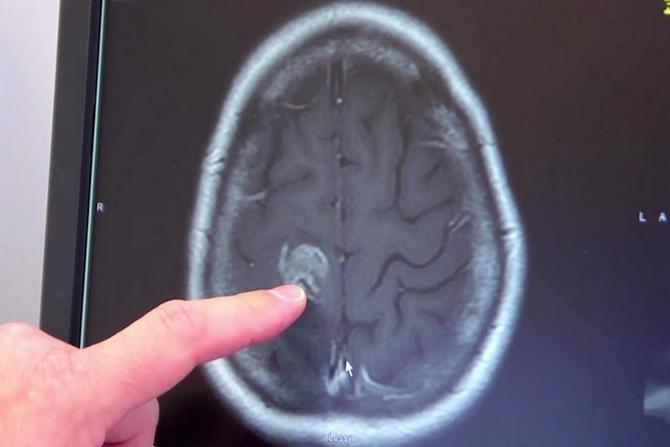
При возникновении признаков заболевания пациенту рекомендуется обратиться к доктору, который проведет сбор анамнеза и осмотр пациента. На основе полученных данных специалист поставит предварительный диагноз. Для его подтверждения рекомендовано проводить компьютерную томографию. Исследование проводится с использованием специальной аппаратуры, что позволяет получить трехмерное изображение позвонков шеи и затылка.
Для того чтобы определить сирингомиелические кисты или менингоцеле рекомендовано проведение магниторезонансной томографии позвоночника. С помощью диагностической процедуры проводится определение не только аномалии, но и других болезней, которые протекают в мозге.
Диагностика заболевания должна быть комплексной, что позволяет определить его тип и разработать эффективную схему лечения.
Лечение
При выявлении аномалии необходимо срочно начать лечение. Заболевание первых двух типов поддается терапии.
Для назначения курса лечения, врач должен изучить все показатели обследования пациента. Если основной признак болезни — головная боль, то будет назначено консервативное лечение.
Будут прописаны нестероидные противовоспалительные препараты, миорелаксанты:
- Ибупрофен;
- Пироксикам;
- Диклофенак;
- Векуроний;
- Атракурий;
- Тубокурарин.
Курс терапии длится несколько месяцев, после чего необходимо пройти полное обследование. При отсутствии положительных результатов назначают хирургическое лечение.
Если консервативная терапия не принесла положительных результатов или у пациента появились серьезные симптомы, то ему назначают ламинэктомию, декомпрессивную краниоэктомию ЗЧЯ. А также пластику твердой оболочки мозга. Данная операция позволяет увеличить объем черепной ямки и расширить затылочное отверстие. Все это приведет к прекращению сдавливания, улучшению оттока цереброспинальной жидкости.
Если имеется гидроцефалия, то производится отведение желудочка при помощи специального клапана.
При проведении операции используется электрофиологический контроль, с помощью которого можно решить открывать полностью твердую оболочку мозга или нет.
Курс реабилитации после хирургического вмешательства занимает семь дней.
Мальформация Киари полностью не изучена и нет способа полностью предотвратить развитие этого заболевания. Но будущие родители, и особенно мамочки могут по максимуму снизить риск возникновения патологии. Для этого необходимо вести правильный образ жизни, изучить все перечисленные выше факторы риска и исключить их по возможности.
Здоровье вашего ребенка зависит от Вас.
19.09.2016
При определенных условиях малыши появляются на свет с врожденными заболевания. Аномалия Арнольда-Киари – заболевания головного мозга, которое связано с нарушениями функций мозжечка, продолговатого мозга, существует несколько вариантов, имеет специфические симптомы, которые сопровождают человека всю жизнь или развиваются со временем. Синдром Арнольда-Киари у плода или взрослого человека влияет на работу сосудодвигательного, дыхательного центра.

Для устранения синдрома Арнольда-Киари применяются обезболивающие и противовоспалительные препараты, а также миорелаксанты
Лечение недуга зависит от степени его проявлений:
- Если синдром протекает бессимптомно и был выявлен случайно, то специфического лечения данной патологии не требуется.
- При наличии у пациента болей в области затылка и шеи применяется консервативная терапия (прием противовоспалительных препаратов, анальгетиков и миорелаксантов).
- При наличии нарушений неврологического характера, которые невозможно устранить консервативно, показано хирургическое вмешательство. Речь идет о проблемах с чувствительностью, парезах, расстройствах мышечного тонуса и т. д.
Для устранения синдрома Арнольда-Киари используются следующие хирургические операции:
- Краниовертебральная декомпрессия. Данная операция предполагает удаление небольшого фрагмента затылочной кости. Это позволяет несколько расширить затылочное отверстие. После этого выполняется резекция мозжечковых миндалин и части двух первых шейных позвонков. Такая процедура позволяет нормализовать циркуляцию церебральной жидкости и устранить симптомы патологии.
- Шунтирование. Операции данного типа предполагают дренирование цереброспинальной жидкости из центрального спинномозгового канала. При этом она может отводиться в брюшную либо в грудную полость.
Причины заболевания
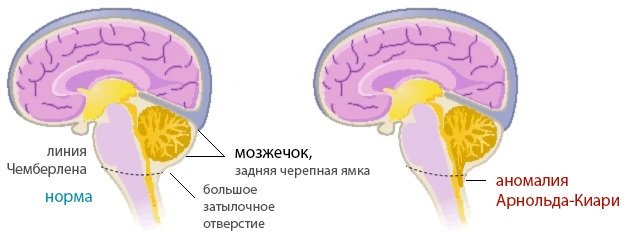
По данным ряда авторов, болезнь Киари представляет собой недоразвитие мозжечка, сочетающееся с различными отклонениями в отделах мозга. Аномалия Арнольда Киари 1 степени – наиболее распространенная форма. Это нарушение представляет собой одностороннее или двухстороннее опускание миндалин мозжечка в спинальный канал. Это может произойти вследствие перемещения продолговатого мозга вниз, часто патология сопровождается различными нарушениями краниовертебральной границы.
Клинические проявления могут возникнуть только на 3–4 десятке жизни. При этом следует отметить, что бессимптомное течение эктопии миндалин мозжечка в лечении не нуждается и часто проявляется случайно на МРТ. На сегодняшний день этиология болезни, так же как и патогенез, изучены плохо. Определенная роль отводится генетическому фактору.
Выделяют три звена в механизме развития:
- генетически обусловленная врожденная остеоневропатия;
- травматизация ската во время родов;
- высокое давление ликвора на стенки спинномозгового канала.
 Болезнь может быть вызвана наследственными нарушениями
Болезнь может быть вызвана наследственными нарушениями
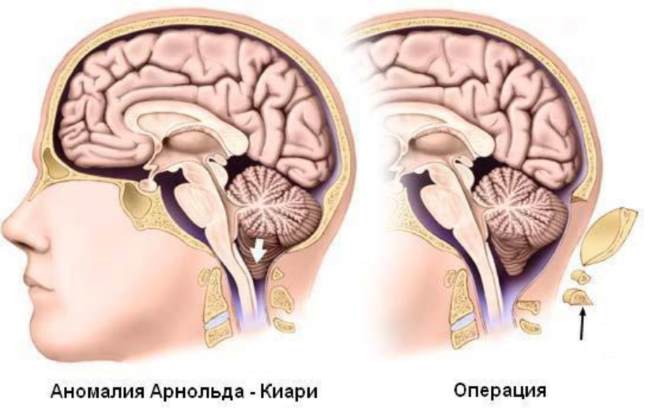
Хирургическое лечение аномалии Арнольда-Киари
Хирургическая коррекция аномалии Арнольда-Киари направлена на расширение костного канала, сдавливающего мягкие образования и устранение ликвородинамических нарушений. Сложность ее состоит в трудной доступности операционного поля, малой величине и высокой плотности расположения жизненно важных нервных центров и проводящих путей.
Операции у детей еще более проблематичны в силу малых размеров анатомических образований и незрелости систем организма. Поэтому они выполняются в условиях нейрохирургических стационаров, оснащенных самой современной операционной техникой и оборудованием для реабилитации послеоперационных больных. С детьми работают высококвалифицированные нейрохирурги, имеющие большой опыт подобных операций.
Современная нейрохирургическая тактика в отношении мальформации Арнольда-Киари предполагает выполнение операций краниовертебральной декомпрессии и шунтирования ликворопроводящей системы. В крупных клиниках развитых стран такие операции выполняются в сопровождении нейронавигации, с применением эндоскопической и микрохирургической аппаратуры. Это позволяет уменьшить тяжесть операционной травмы и предотвратить случайное повреждение важных областей.
Кроме первичных, нейрохирургам ведущих зарубежных центров нередко приходится выполнять ревизионные операции маленьким иностранным пациентам после неудачных вмешательств, выполненных у них на родине. Проводится:
Краниовертебральная декомпрессия. Под общим наркозом удаляют часть затылочной кости, расширяя большое затылочное отверстие, а также задние части верхних шейных позвонков (ламинэктомия) до уровня провисания ткани мозга. Кроме того, делают резекцию опустившихся миндалин мозжечка, тем самым устраняя давление на ствол головного мозга и верхние отделы спинного мозга.
В ходе операции вскрывается и твердая мозговая оболочка. В разрез вшивают для облегчения циркуляции спинномозговой жидкости «заплату» из синтетического материала или собственных тканей пациента, например, фасции бедра.
Шунтирование ликворопроводящих путей. Поскольку циркуляция спинномозговой жидкости при аномалии Арнольда-Киари на уровне краниовертебрального перехода затруднена, часть ее перенаправляют через имплантированные шунты (синтетические трубочки) в грудную клетку или брюшную полость. Там ликвор всасывается, и внутричерепное давление снижается.
Причины болезни
На данный момент причины развития заболевания не до конца изучены. Существует ряд провоцирующих факторов, при воздействии которых развивается заболевание. Если женщина при беременности бесконтрольно принимает медикаментозные препараты, то это приводит к патологии. Она развивается, если при вынашивании женщина курит или регулярно принимает спиртные напитки. Если во время беременности протекают заболевания вирусного характера, то это повышает риск развития болезни.

Мальформация Киари диагностируется при врожденных патологиях – чрезмерно маленьких размерах задней черепной ямки или большом затылочном отверстии. У взрослых пациентов патологический процесс появляется после травм черепа. Синдром возникает при гидроцефалии.
Существует большое количество причин возникновения болезни, которые рекомендуется устранять из жизни.
Причины и факторы риска
Исследователи полагают, что синдром Арнольда-Киари может иметь наследственное происхождение, так как обнаруживалась среди членов одной семьи. Тем не менее, генетические условия, вызывающие заболевание (т.е., какие и сколько генов участвуют) и тип передачи еще предстоит выяснить.
Исходя из серьезности выпячивания и момента жизни, в котором он возникает, заболевание можно разделить на 4 различных типа, идентифицированных первыми четырьмя римскими числами (I, II, III и IV).
Первые два типа по сравнению со вторыми более распространены и менее серьезны; Тип III и тип IV, на самом деле, очень редки и несовместимы с жизнью.
— Мальформация I типа.
Первая степень синдрома протекает бессимптомно (т.е. без явных симптомов), по крайней мере, до конца детства или юности.
Причина его возникновения кроется в уменьшенном черепном пространстве: в таких условиях часть мозжечка (именно миндалина(и), расположенная(ые) с нижней стороны), из-за недостатка места, вынуждена проникать в затылочное отверстие и входить в позвоночный канал.
Примечание: у некоторых взрослых людей с синдром Арнольда-Киари 1 типа все в порядке и они ведут совершенно нормальную жизнь. Это связано с тем, что аномалия мозжечка не настолько серьезна, чтобы вызывать симптомы или нарушения. Поэтому очень часто эти субъекты игнорируют свое состояние или узнают о нем по чистой случайности.
— Мальформация II типа.
2 тип мальформации Арнольд-Киари является врожденным заболеванием, которое присутствует с рождения ребенка, и всегда протекает симптоматически.
По сравнению с 1 степенью он характеризуется большим выпячиванием черепной ямки, при котором помимо миндалин мозжечка также выпячивает часть мозжечка (называемая червь мозжечка) и венозный сосуд.
Почти всегда мальформация Арнольд-Киари II типа ассоциируется с особой формой расщелины позвоночника, называемой миеломенингоцеле.
Среди различных последствий этой аномалии выделают: блокирование потока ликвора (спинномозговой жидкости) через затылочное отверстие (что приводит к состоянию, называемому гидроцефалией) и прерывание нервных сигналов.
— Мальформация III типа.
Присутствующий с рождения, III тип порока вызывает серьезные неврологические проблемы, настолько, что часто несовместимы с жизнью. В этих случаях на самом деле наблюдается выпячивание мозжечка, и по этой причине говорится о затылочном энцефалоцеле.
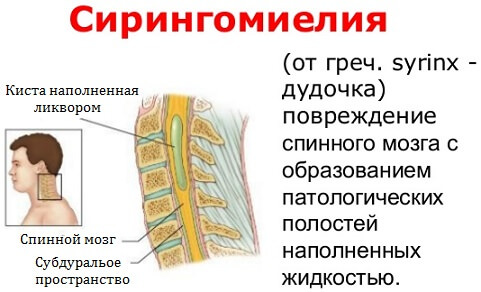
Обычно III тип состояния характеризуется гидроцефалией и сирингомиелией; последний представляет собой особое состояние, характеризующееся наличием одной или нескольких кист в позвоночном канале.
— Мальформация IV типа.
Мальформация Арнольда-Киари IV типа характеризуется отсутствием развития части мозжечка (недоразвитие мозжечка).
Аномалия врожденная и абсолютно несовместима с жизнью.
Связанные расстройства
Врачи и ученые отметили, что следующие заболевания являются частыми среди людей с мальформацией Киари:
- гидроцефалия;
- сирингомиелия;
- сколиоз;
- синдром Марфана;
- синдром Элерса-Данлоса.
Эпидемиология
Точная частота пороков развития неизвестна; это связано с тем, что у некоторых даже взрослых людей с I типом мальформации Арнольда-Киари никаких симптомов нет, и они кажутся совершенно нормальными (поэтому болезнь недиагностируется).
Несколько достоверных эпидемиологических исследований сообщают, что:
- I тип симптоматичен у 1 из 100 детей;
- II тип особенно широко распространен в популяциях кельтского происхождения;
- женщины страдают в 3 раза чаще, чем мужчины.
Диагностика и лечение мальформации Арнольда-Киари
Диагностикой этого состояния занимается невролог и нейрохирург. Кроме внешнего осмотра и оценки жалоб пациента, специалист назначает такие исследования, как:
- ЭХО-ЭГ;
- ЭЭГ;
- рентгенография.
Для подтверждения диагноза нередко назначается МРТ головного и спинного мозга.
Лечение
После постановки диагноза может назначаться консервативное или оперативное лечение. Применять народные рецепты и средства китайской медицины нецелесообразно. Если симптомы патологии проявляются слабо, для их коррекции могут назначаться нестероидные противовоспалительные средства и миорелаксанты.
Если признаки аномалии развития имеют выраженный характер, требуется хирургическое вмешательство. Нередко выполняется операция по трепанации задней черепной коробки и удалению части затылочной кости. Может потребоваться резекция части миндалевидного тела и спаек.
Осложнения
При 1 типе синдрома Арнольда-Киари тяжелые последствия течения аномалии нередко отсутствуют. Если терапия проводилась консервативными методами и произошло повышение внутричерепного давления на фоне травмы или других нарушений, возможно развитие паралича конечностей.
Если подобное нарушение имеет беременная женщина, ей требуется особое внимание со стороны врачей в период родовой деятельности, т. к
повышение внутричерепного давления во время натуживания может стать причиной развития ишемического инсульта спинного или головного мозга. Если терапия проводилась хирургическими методами, возможно ухудшение оттока ликвора, неврологические нарушения и летальный исход.
Факторы риска
К факторам риска, ухудшающим прогноз лечения, относятся артериальная гипертензия и заболевания органов дыхания, ожирение и присутствующие ишемические повреждения тканей спинного и головного мозга.
Прогноз
При своевременном проведенном комплексном лечении синдром Арнольда-Киари 1 и 2 типов не оказывает влияние на продолжительность жизни больного. При 3 и 4 типах заболевания велика вероятность преждевременной гибели больного.
Причины
Существует несколько основных версий о том, почему появляется церебральное заболевание Арнольда Киари. У каждой есть свои сторонники. Так, ранее аномалию относили исключительно к врожденным мальформациям. Однако, практика специалистов это опровергла – порок у отдельных больных был приобретен в результате событий их жизни.
К внутриутробному формированию дефекта мозговых структур приводят:
- злоупотребления женщиной алкогольной продукцией;
- бесконтрольный прием медикаментов, особенно на первом этапе беременности;
- перенесенные будущей матерью инфекционные и вирусные заболевания – к примеру, цитомегаловирус, краснуха, ветрянка.
Приобретенная аномалия Арнольда, как правило, – это результат несоответствия между размерами костных структур черепа в районе задней черепной ямки и находящихся в ней мозговых отделов. К подобному состоянию приводят различные травмы – родовые, автодорожные, бытовые.
В ряде ситуаций аномалия будет последствием гидроцефалии – возрастание размеров головного мозга за счет избытка продуцирования либо сбоя оттока ликвора. Это постепенно приводит к росту внутричерепного давления и дальнейшему смещению структур по направлению к затылочному кольцу.
Общая информация
Синдром Арнольда-Киари представляет собой набор признаков и симптомов, вызванных редкой мальформацией (отклонение от нормального развития, аномалия) задней черепной ямки; у пострадавших эта структура развита слабо, поэтому мозжечок выходит (выступает) из своего естественного участка через затылочное отверстие, расположенное у основания черепа.
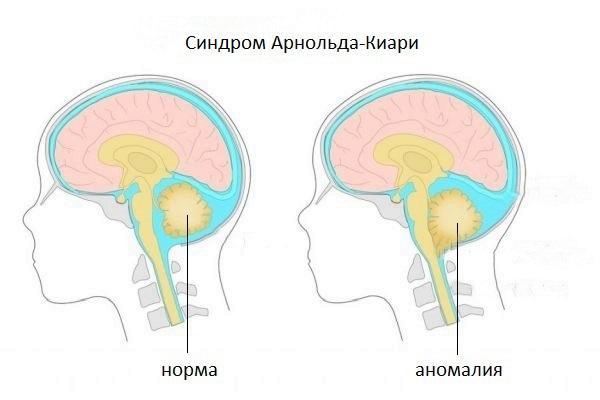
Есть четыре различных типа синдрома Арнольда-Киари; особенность, отличающая один тип от другого, является степень выпячивания, следовательно, доля вовлеченного материала мозжечка. Тип I является наименее тяжелым (иногда остается бессимптомным на протяжении всей жизни), тогда как IV тип наиболее тяжелый; однако уже со второго типа качество жизни больного ставится под угрозу.
Симптомы, характеризующие аномалии Арнольда-Киари многочисленны и варьируются от головных болей до слабости мышц и проч.
На сегодняшний день не существует лекарств, позволяющих устранить порок развития мозжечка, однако существуют способы лечения, позволяющие частично смягчить симптомы.