Талассемия
Содержание:
- Проблемы терапии
- Лечение талассемии
- Как жить с талассемией?
- Разновидности заболевания при нарушении бета-цепочек
- Диагностирование заболевания
- Варианты и формы
- Что такое эритроциты?
- Что такое талассемия?
- Разновидности альфа-талассемии
- Классификация [ править | править код ]
- Прогноз для больных талассемией
- Медицинские статьи
- Классификация
- Группы риска
- Диагностика
- Почему происходит разрушение эритроцитов
- Варианты и формы
- Online-консультации врачей
- Прогноз течения болезни
- Клинические проявления бета-талассемии
- Разновидности бета-талассемии
Проблемы терапии
Лечение талассемии зависит от тяжести поражения эритропоэза, степени охвата генов процессами мутации. В настоящее время применяются следующие методы.
Диетический рацион направлен на снижение всасывания железа в кишечнике, рекомендуются орехи, какао, соя, чай.
Тяжелая форма требует регулярного переливания крови, эритроцитарной массы, размороженных и профильтрованных эритроцитов. Эффективность временная, возможны побочные эффекты, но главное — сохранить жизнь больному.
Терапия дополняется ежедневным устранением излишков железа с помощью введения (хелатов — специальных комплексов, повышающих действие лекарства). Для связывания железа назначается Десферал. Этот препарат предупреждает сидероз (патологическое состояние, вызванное отложением в тканях железа), но на уровень гемоглобина не влияет.
При возникновении резкого ухудшения состояния по аналогии с гемолитическими кризами показаны глюкокортикоиды в больших дозах.
Спленэктомия возможна при больших размерах селезенки детям после пятилетнего возраста. Наиболее оптимален возраст 8–10 лет. После удаления селезенки наступает период улучшения, но опасен риск присоединения инфекции.
Для трансплантации необходим донор, совпадающий по всем параметрам, лучше всего из близких родственников.
К симптоматическим средствам относятся препараты гепатопротекторного действия, большие дозы аскорбинки помогают вывести излишки железа из организма.
Все формы талассемии нуждаются в препаратах с фолиевой кислотой и витаминами группы В. На фоне присоединившейся инфекции, при беременности следует применять фолиевую кислоту в больших дозах, поскольку неэффективное кроветворения при талассемии значительно увеличивает ее потребление клетками.
Лечение талассемии
Терапевтические меры зависят от:
- Степени тяжести эритропоэза;
- Степени поражения генов мутационным процессом.
К главным методам лечения талассемии принадлежит:
- Диета. Ее задача — снизить всасывание Fe кишечником. Для этого в меню вводят орехи, соевые продукты, чай/какао.
- Введение хелатов. Эти средства повышают активность основных лекарств, а также ликвидируют переизбыток Fe. Применяются ежедневно.
- Прием «Десферала». Этот препарат связывает Fe, тем самым предотвращает его застаивание в тканях. На уровень гемоглобина лекарство не влияет.
- Переливание крови, эритроцитов (профильтрованных и размороженных), эритроцитарной массы. Метод используется при тяжелой форме заболевания, когда необходимо сохранить пациенту жизнь.
- Прием больших доз глюкокортикоидов. Они уместны, когда развивается гемолитический криз.
- Удаление селезенки. Операция назначается при увеличенной селезенке. Спленэктомия производится с 5 лет, однако приемлемым возрастом считается 8-10 лет. Улучшение после операции наступает, но велик риск присоединения инфекции.
Препараты симптоматического действия, необходимые для лечения талассемии
- Гепатопротекторы;
- Аскорбиновая кислота (выводит излишек Fe);
- Средства на основе фолиевой кислоты;
- Витамины группы В.
Как жить с талассемией?
Хотя вы никак не можете предотвратить наследование талассемии, вы способны при помощи определенных действий максимально приблизить качество жизни к оптимальному. К таковым относятся:
- Четкое следование плану лечения. Делайте переливания крови настолько часто, насколько рекомендует врач, принимайте фолиевую кислоту и/или проходите хелатирующую терапию.
- Беспрерывное лечение. Регулярно посещайте врача для прохождения осмотров и сдавайте все назначенные им анализы. Это могут быть как специализированные тесты, относящиеся к талассемии, так и индикаторы общего состояния здоровья. Не забывайте прививаться от гриппа, пневмонии, гепатита В и менингита.
- Здоровый образ жизни. Питайтесь согласно составленному плану. Во время пика сезонных заболеваний регулярно мойте руки и избегайте скоплений людей во избежание инфицирования. Следите за чистотой в помещении, особенно в местах проведения переливания. При появлении жара или других признаков инфекции сразу же обратитесь к врачу.
- Группы поддержки. Присоединитесь к существующим в вашем регионе группам поддержки, чтобы делиться с другими людьми опытом жизни с талассемией. Но ни в коем случае не вносите коррективы в план лечения без предварительного одобрения со стороны лечащего врача.
Разновидности заболевания при нарушении бета-цепочек
Гены, которые контролируют выработку бета-цепочек, могут находиться в разном состоянии:
- Нормальный здоровый ген. У здоровых людей он находится в норме, что обеспечивает выработку правильных цепочек гемоглобина.
- Ген с частичными отклонениями. Такая патология позволяет организму вырабатывать нормальный гемоглобин, однако, он продуцируется в недостаточном количестве.
- Ген искажен настолько, что полностью препятствует формированию бета-цепей.
В зависимости от масштабов заболевания различают следующие виды бета-талассемии:
- Минорная талассемия. При таком виде заболевания поврежден только один ген. Симптоматика практически отсутствует. Может наблюдаться незначительная анемия. Помимо малокровия, человек не жалуется на состояние своего здоровья.
- При более серьезной патологии гена, отвечающего за синтез бета-цепей, наблюдается талассемия интермедиа. В результате нарушения процесса производства гемоглобина эритроциты формируются либо недоразвитыми, либо маленьких размеров. Переливания крови не являются необходимостью, однако, последующее состояние организма напрямую зависит от его способности жить при низком уровне гемоглобина.
- Бета-талассемию типа «майор» характеризует нарушением всех генов, отвечающих за синтез бета-цепей. Больные таким типом заболевания требуют регулярных переливаний крови. Такая процедура позволяет поддерживать жизнь пациента.
Диагностирование заболевания
Выявить недуг можно даже при отсутствии клинической картины
Медицина предусматривает полный комплекс принципов диагностики, предназначенных для обнаружения у человека бета-талассемии. Выявить недуг можно даже при отсутствии клинической картины. Основой для установления диагноза служат:
- Присутствие особых патологий, разрушающих эритроциты.
- Нарушение обычного строения эритроцитов.
- Наследственное происхождение патологии.
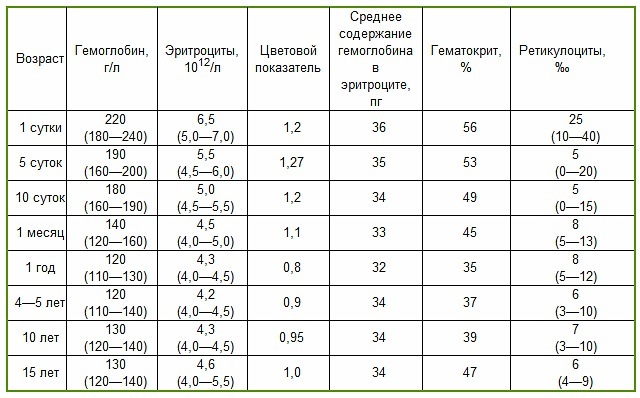
При диагностике доктору требуются сведения о состоянии здоровья родителей больного для обнаружения вероятности генетически спровоцированных мутаций. Для этого также выполняется исследование крови на ДНК. Главным лабораторным способом установления диагноза талассемия становятся биохимические показатели крови, а именно:
- снижение показателей Hb до 50 г/л при гомозитном виде и до 110 г/л при гетерозиготном виде недуга;
- цветовой показатель не достигает 0,5;
- рост предшественников эритроцитов составляет менее 4%.
Тесты биохимии отражают сбои в обмене железа, как и в случае гемолитической анемии:
- повышенная степень свободного элемента билирубина;
- высокое содержание железа в сыворотке;
- пониженная возможность к связыванию железа.
Кроме лабораторных анализов крови проводится ряд исследований другого типа. Пункция костного мозга помогает выявить объем кроветворного ростка, краниограмма позволяет определить игольчатый периостоз. Ультразвуковое исследование брюшной полости способствует фиксированию измененных объемов печени и селезенки. При наличии подозрений на бета-талассемию докторам необходимо исключить различные типы анемий.
Варианты и формы
Люди, унаследовавшие аномальный гемоглобин от отца и от матери, почти всегда обречены на страдания по причине тяжелой формы болезни, которую называют большой талассемией (thalassaemia major) или болезнью Кули. Лица, получившие подобный «подарок» только от кого-то одного, имеют возможность прожить жизнь и не заметить свой недуг, если к нему не приведут неблагоприятные обстоятельства. Это значит, что бета-талассемия имеет различные формы:
-
- Гомозиготная β-талассемия, полученная от обоих родителей и названная в честь врача, описавшего ее, болезнью Кули, характеризуется значительным увеличением содержания фетального (HbF) красного пигмента (до 90%), проявляется у детей где-то к концу первого года жизни и отличается обилием симптомов;
- Гетерозиготная форма, которую называют малой талассемией, является результатом наследования патологии только от одного из родителей, поэтому признаки малокровия большей частью стерты, а в некоторых случаях какие бы то ни было симптомы болезни и вовсе отсутствуют.
Однако такого разделения можно придерживаться при классическом варианте течения. На деле все не столь однозначно. У некоторых больных с гомозиготной формой отсутствуют явные признаки тяжелой патологии, и анемия не настолько выражена, чтобы заставлять больного жить на постоянных гемотрансфузиях, поэтому такую талассемию, не глядя на гомозиготность, нельзя назвать большой – ее квалифицируют как промежуточную.
Кроме этого, в течении гомозиготной талассемии различают 3 степени тяжести:
-
- Тяжелая гомозиготная талассемия, диагностируемая только у детей первого года жизни, поскольку больше прожить эти малыши просто не могут по причине тех изменений, которые происходят в организме;
- Среднетяжелая талассемия хоть и протекает в менее тяжелой форме, но все же не позволяет малышу перейти 8-летний возраст;
- Наиболее легкая форма (назвать просто «легкой» как-то не получается) – она дает шанс перешагнуть подростковый возраст и вступить во взрослую жизнь, чтобы потом эту жизнь в самом расцвете забрать.
Вместе с тем, отдельные случаи гетерозиготных бета-талассемий протекают в более тяжелой форме, нежели от них ждут. Такое несоответствие объясняется неспособностью красных кровяных телец избавляться от лишних альфа-цепей, хотя для гетерозиготной формы болезни, в принципе, свойственна повышенная утилизация ненужных цепей. В связи с этим, возникает необходимость делить болезнь на формы в зависимости от степени выраженности клинических проявлений вне зависимости от гомо- или гетерозиготности. И различать:
-
- Большую талассемию;
- Промежуточную форму;
- Малую талассемию;
- Минимальный вариант.
Однако такое деление интересно специалистам, читателю, скорее всего, хочется знать об основных признаках классических вариантов гомозиготных и гетерозиготных талассемий.
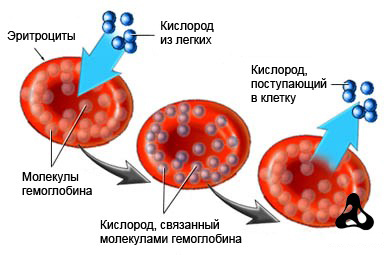
Что такое эритроциты?
кислорода и углекислого газа
Образование эритроцитов
эритропоэзОсновными кроветворными органами являются:
- Печень. Первые очаги кроветворения в печени появляются на 6 неделе внутриутробного развития. Начиная с этого периода и до рождения ребенка печень является основным местом образования клеток крови. После рождения кроветворная функция печени угнетается.
- Селезенка. Развитие селезенки начинается на 5 – 6 неделе беременности, однако первые очаги кроветворения в ней появляются на 4 месяце внутриутробного развития. Кроветворная функция селезенки, как и печени, угнетается после рождения ребенка.
- Красный костный мозг (ККМ). Это особое вещество, располагающееся в губчатых костях организма (в костях таза, черепа, позвонках, грудине), а также в длинных трубчатых костях (плеча и предплечья, бедра и голени). Образование эритроцитов в ККМ начинается с 4 месяца беременности. После рождения ребенка, когда кроветворение в печении и селезенке угнетается, красный костный мозг становится единственным кроветворным органом человека.
Из стволовой клетки крови развиваются:
- Эритроциты – обеспечивают транспорт газов в организме.
- Тромбоциты – ответственны за остановку кровотечений.
- Лейкоциты – защищают организм от различных инфекций (бактерий, вирусов и т. д.).
- Лимфоциты – обеспечивают и регулируют защитные функции организма.
размножениянедостаток кислородапревращения стволовой клетки в эритроцитпоследовательно образуются проэритробласт, эритробласт, нормобластмолодая форма эритроцитаэто обусловлено наличием в нем красного пигмента гема
Образование гемоглобина
белка30 пикограммДНКдезоксирибонуклеиновой кислотыпо 2 от каждого родителяпо 1 от каждого родителягемВ зависимости от периодов развития различают:
- Эмбриональный гемоглобин (HbU). Встречается в организме эмбриона с 3 по 10 недели внутриутробного развития.
- Фетальный гемоглобин (HbF). Является основным типом гемоглобина плода. Его количество постепенно уменьшается, и в период рождения составляет только 20% от общего гемоглобина организма. У взрослого человека встречается в незначительных количествах.
- Взрослый гемоглобин (HbA). При рождении ребенка 80% всего гемоглобина представлено HbA, а к возрасту 2 – 3 лет и до конца жизни его доля составляет 98%.
- Малый компонент взрослого гемоглобина (HbA2). Составляет примерно 2% от общего гемоглобина взрослого человека.
Что такое талассемия?
Красный кровяной пигмент (гемоглобин, Hb) представляет собой один из главных компонентов красных кровяных телец – эритроцитов, обеспечивающих выполнение такой важной функции, как транспортировка кислорода (О2) от легких к тканям и наоборот – забирая углекислый газ из тканей, Hb участвует в выведении продуктов обмена из организма. Химическое строение молекулы гемоглобина самым тесным образом связано с его биологическими задачами, ведь именно сложная структура молекулы Hb обеспечивает выполнение физиологических функций, которые заключаются в переносе кислорода и питании тканей
Разумеется, эритроциты, несущие аномальный гемоглобин, теряют способность к качественному выполнению своей работы, что приводит к развитию у человека гипохромной анемии.
2+
Бета-талассемию, в отличие от альфа-талассемии, когда происходит выпадение генов альфа-цепей, обычно не связывают с делецией (deletion- уничтожение) генов. Дефект преимущественно формируется за счет образования неполноценной м-РНК и бета-глобина, вследствие чего падает продукция бета-цепей.
Разновидности альфа-талассемии
В зависимости от степени мутации гена эту форму заболевания делят на несколько групп:
- Происходит мутация одного локуса гена. В этом случае можно и не наблюдать клинических проявлений.
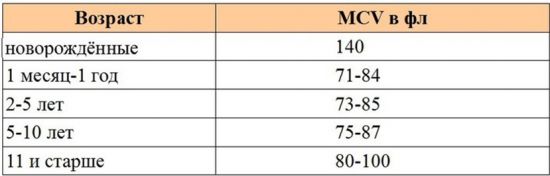
- Поражение происходит в двух локусах, причем они могут находиться на одном гене или на разных. В анализе крови хорошо диагностируется низкий уровень гемоглобина и маленькие эритроциты.
- Три локуса в генах подвержены мутации. Нарушается перенос кислорода к тканям и органам. В некоторых случаях еще происходит увеличение селезенки.
- Мутация во всех локусах приводит к полному отсутствию синтеза альфа-цепей. С такой формой гибель плода происходит еще внутри утробы матери или сразу после рождения.
Если легкой формы альфа-талассемия, лечение может и не понадобиться, а вот с тяжелой разновидностью придется всю жизнь находиться под контролем врачей. Только регулярные курсы терапии могут повысить качество жизни человека.
Классификация [ править | править код ]
В зависимости от того, синтез какого из мономеров нарушен, разделяют альфа-, бета- и дельта-талассемию. По тяжести клинических проявлений выделяют тяжёлую, среднюю и лёгкую формы заболевания.
Альфа-талассемия
Связана с мутациями в генах HBA1 и HBA2. Есть всего 4 локуса, кодирующего α-цепи. Наличие мутации в одном из локусов приводит к минимальным клиническим проявлениям. Нарушения в двух локусах выражаются лёгкой формой анемии. При мутациях в трёх локусах возникает значительное уменьшение продукции α-глобина. При этом избыточные цепи β-глобина образуют тетрамеры — гемоглобин Н. Эта форма носит также название гемоглобинопатии Н. Характер заболевания может варьироваться от лёгкой до тяжёлой картины гипохромной микроцитарной анемии. Присутствие мутаций во всех четырёх аллелях альфа-глобина не совместимо с жизнью. Ребёнок с такой патологией погибает внутриутробно или вскоре после рождения. Из пуповинной крови таких детей можно выделить гемоглобин Барта.
Бета-талассемия
Существует два варианта бета-талассемии — большая талассемия CD8(-AA) и малая талассемия (minor), из которых большая талассемия — наиболее тяжёлая форма заболевания. Возникает при наличии мутаций в обоих аллелях гена бета-глобина. В отсутствие или при резком уменьшении производства бета-цепей гемоглобин А вытесняется гемоглобином F, в норме вырабатывающимся у плода и сменяющимся на гемоглобин А после родов. Малая талассемия связана с наличием мутации в одном из аллелей гена бета-глобина. Как правило, протекает легко и не требует лечения.
Прогноз для больных талассемией
В зависимости от тяжести и формы заболевания прогноз может быть различным. С малой талассемией больные живут нормальной жизнью, и продолжительность ее практически не отличается от продолжительности жизни здоровых людей.
При бета-талассемии малая часть пациентов доживает до половозрелого возраста.
Гетерозиготная форма заболевания практически не требует лечения, а вот при гомозиготной, да еще и тяжелой формы, необходимо делать регулярное переливание крови. Без этой процедуры жизнь пациента практически невозможна.
К большому сожалению, талассемия в настоящее время относится к тем заболеваниям, с которыми наука еще не научилась справляться. Можно только в какой-то степени держать ее под контролем.
Медицинские статьи
Офтальмология является одной из наиболее динамично развивающихся областей медицины. Ежегодно появляются технологии и процедуры, позволяющие получать результат, который еще 5–10 лет назад казался недостижимым. К примеру, в начале XXI века лечение возрастной дальнозоркости было невозможно. Максимум, на что мог рассчитывать пожилой пациент, — это на…
Почти 5% всех злокачественных опухолей составляют саркомы. Они отличаются высокой агрессивностью, быстрым распространением гематогенным путем и склонностью к рецидивам после лечения. Некоторые саркомы развиваются годами, ничем себя не проявляя…
Вирусы не только витают в воздухе, но и могут попадать на поручни, сидения и другие поверхности, при этом сохраняя свою активность. Поэтому в поездках или общественных местах желательно не только исключить общение с окружающими людьми, но и избегать…
Вернуть хорошее зрение и навсегда распрощаться с очками и контактными линзами – мечта многих людей. Сейчас её можно сделать реальностью быстро и безопасно. Новые возможности лазерной коррекции зрения открывает полностью бесконтактная методика Фемто-ЛАСИК.
Классификация
В зависимости от того, синтез какого из мономеров нарушен, разделяют альфа-, бета- и дельта-талассемию.
По тяжести клинических проявлений выделяют тяжёлую, среднюю и лёгкую формы заболевания.
Альфа-талассемия
Связана с мутациями в генах HBA1 и HBA2. Есть всего 4 локуса, кодирующего α-цепи. Наличие мутации в одном из локусов приводит к минимальным клиническим проявлениям. Нарушения в двух локусах выражаются лёгкой формой анемии. При мутациях в трёх локусах возникает значительное уменьшение продукции α-глобина. При этом избыточные цепи β-глобина образуют тетрамеры — гемоглобин Н. Эта форма носит также название гемоглобинопатии Н. Характер заболевания может варьироваться от лёгкой до тяжёлой картины гипохромной микроцитарной анемии. Присутствие мутаций во всех четырёх аллелях альфа-глобина не совместимо с жизнью. Ребёнок с такой патологией погибает внутриутробно или вскоре после рождения. Из пуповинной крови таких детей можно выделить гемоглобин Барта.
Бета-талассемия
Существует два варианта бета-талассемии — большая талассемия CD8(-AA) и малая талассемия (minor), из которых большая талассемия — наиболее тяжёлая форма заболевания. Возникает при наличии мутаций в обоих аллелях гена бета-глобина. В отсутствие или при резком уменьшении производства бета-цепей гемоглобин А вытесняется гемоглобином F, в норме вырабатывающимся у плода и сменяющимся на гемоглобин А после родов.
Малая талассемия связана с наличием мутации в одном из аллелей гена бета-глобина. Как правило, протекает легко и не требует лечения.
Группы риска
Частота возникновения альфа талассемии среди белых людей низкая. Около 15% афроамериканцев являются её носителями, и только 3% имеют слабо выраженные признаки. Гемоглобинопатия H редко встречается в этой группе населения. В Северной Америке из-за присутствия большого количества мультинациональных слоёв общества частота заболевания синдромом альфа талассемии увеличилась. У некоторых этнических групп (население Юго-Восточной Азии и средиземноморского населения) гемоглобинопатия H встречается чаще. Высокая частота проявления гемоглобинопатии Constant Spring (CS) была отмечена в Юго-Восточной Азии.
Наследственность
Это заболевание наследуется как не полностью доминантный аутосомный признак от родителей к детям через мутировавшие гены гемоглобина.
Если в семье присутствовали случаи талассемии, ребенок имеет повышенный риск развития заболевания.
Международная статистика
Альфа-талассемия – наиболее распространённое генетическое заболевание в мире. Существует 270 миллионов носителей мутировавших генов глобина, которые могут передать тяжелые формы талассемии по наследству. Кроме того, 300000-400000 больных младенцев рождаются каждый год, более 95% из которых находятся в Азии, Индии или на Ближнем Востоке.
Частота проявления этой болезни составляет 20-30% в частях Западной Африки, 60-80% — в некоторых районах Саудовской Аравии, Индии, Таиланда, Папуа-Новой Гвинеи и Меланезии. В Таиланде около 7000 детей ежегодно рождается с гемоглобинопатией H. Среди китайского населения диапазон больных составил от 5% до 15%. Частота возникновения альфа-талассемии в Великобритании, Исландии и Японии ниже 0,01%.
Пол
Мужчины и женщины затронуты в равной степени.
| Талассемия | |
|---|---|
| МКБ-10 | D 56 56. |
| МКБ-10-КМ | D56 и D56.9 |
| МКБ-9 | 282.4 282.4 |
| МКБ-9-КМ | 282.4 , 282.40 и 282.49 |
| MedlinePlus | 000587 |
| eMedicine | ped/2229 |
| MeSH | D013789 и D013789 |
Талассеми́я (анемия Кули) — заболевание, наследуемое по рецессивному типу (двухаллельная система), в основе которого лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина. В норме основным вариантом (97 %) гемоглобина взрослого человека является гемоглобин А. Это тетрамер, состоящий из двух мономеров α-цепей и двух мономеров β-цепей. 3 % гемоглобина взрослых представлено гемоглобином А2, состоящим из двух альфа- и двух дельта-цепей. Существуют два гена HBA1 и HBA2, кодирующих мономер альфа, и один HBB-ген, кодирующий мономер бета. Наличие мутации в генах гемоглобина может привести к нарушению синтеза цепей определённого вида.
Диагностика
При диагностике талассемии берутся во внимание данные клинических симптомов и анамнез. Обязательно у пациента берется кровь на обследование
Малокровие будет заметно уже в общем анализе. Также будет обнаружен высокий билирубин, сывороточное железо, АЛТ будут за пределами нормы.
В общей сложности диагностические мероприятия выглядят следующим образом:
- Анализ крови на гемоглобин.
- Клинический и биохимический анализ крови.
- Анализ крови общий.
- Пункция костного мозга.
- Генетический тест.
- УЗИ органов брюшной полости.
- Цитогенетическое исследование околоплодных вод.
С целью выявления увеличения выработки железа с мочой, используют такие методики диагностики, как электрофорез гемоглобина и десфераловый тест.
Почему происходит разрушение эритроцитов
Талассемия – это болезнь генетического происхождения, возникающая в случае наследования аномального гена от обоих родителей и его отсутствии в половых хромосомах
Талассемия – это болезнь генетического происхождения, возникающая в случае наследования аномального гена от обоих родителей и его отсутствии в половых хромосомах. Прямым фактором возникновения патологии становятся разнообразные мутационные сбои в гене, кодирующем синтез одной било другой цепи Hb. В основу молекул патологии могут входить синтез дефектной аномальной мРНК, структурное изменение генетического материала, мутация и непродуктивная транскрипция регуляторных генов. Результатом таких патологий становится сокращение либо прекращение синтеза одной из полипептидных гемоглобиновых цепочек.
Таким образом, при бета-талассемии синтез β-цепей происходит в неполном объеме, а это вызывает формирование избыточных α-цепей, и наоборот. Излишне синтезированные полипептидные цепочки сохраняются в клетках эритроцидного типа, повреждая их. Процессу сопутствует разрушение эритрокариоцитов в костном мозге, гемолиз эритроцитов в периферической крови, уничтожением ретикулоцитов в селезенке. Помимо того, при бета-талассемии эритроциты накапливают фетальный Hb, не транспортирующий кислород в ткани, вследствие чего образуется тканевая гипоксия. Костномозговая гиперплазия провоцирует деформирование скелетных костей. Малокровие, гипоксия тканей и неэффективный эритропоэз в большей или меньшей степени вызывают нарушения в росте и развитии ребенка.
Варианты и формы
веряотности наследования талассемии
Люди, унаследовавшие аномальный гемоглобин от отца и от матери, почти всегда обречены на страдания по причине тяжелой формы болезни, которую называют большой талассемией (thalassaemia major) или болезнью Кули. Лица, получившие подобный «подарок» только от кого-то одного, имеют возможность прожить жизнь и не заметить свой недуг, если к нему не приведут неблагоприятные обстоятельства. Это значит, что бета-талассемия имеет различные формы:
- Гомозиготная β-талассемия, полученная от обоих родителей и названная в честь врача, описавшего ее, болезнью Кули, характеризуется значительным увеличением содержания фетального (HbF) красного пигмента (до 90%), проявляется у детей где-то к концу первого года жизни и отличается обилием симптомов;
- Гетерозиготная форма, которую называют малой талассемией, является результатом наследования патологии только от одного из родителей, поэтому признаки малокровия большей частью стерты, а в некоторых случаях какие бы то ни было симптомы болезни и вовсе отсутствуют.
Однако такого разделения можно придерживаться при классическом варианте течения. На деле все не столь однозначно. У некоторых больных с гомозиготной формой отсутствуют явные признаки тяжелой патологии, и анемия не настолько выражена, чтобы заставлять больного жить на постоянных гемотрансфузиях, поэтому такую талассемию, не глядя на гомозиготность, нельзя назвать большой – ее квалифицируют как промежуточную.
Кроме этого, в течении гомозиготной талассемии различают 3 степени тяжести:
- Тяжелая гомозиготная талассемия, диагностируемая только у детей первого года жизни, поскольку больше прожить эти малыши просто не могут по причине тех изменений, которые происходят в организме;
- Среднетяжелая талассемия хоть и протекает в менее тяжелой форме, но все же не позволяет малышу перейти 8-летний возраст;
- Наиболее легкая форма (назвать просто «легкой» как-то не получается) – она дает шанс перешагнуть подростковый возраст и вступить во взрослую жизнь, чтобы потом эту жизнь в самом расцвете забрать.
Вместе с тем, отдельные случаи гетерозиготных бета-талассемий протекают в более тяжелой форме, нежели от них ждут. Такое несоответствие объясняется неспособностью красных кровяных телец избавляться от лишних альфа-цепей, хотя для гетерозиготной формы болезни, в принципе, свойственна повышенная утилизация ненужных цепей. В связи с этим, возникает необходимость делить болезнь на формы в зависимости от степени выраженности клинических проявлений вне зависимости от гомо- или гетерозиготности. И различать:
- Большую талассемию;
- Промежуточную форму;
- Малую талассемию;
- Минимальный вариант.
Однако такое деление интересно специалистам, читателю, скорее всего, хочется знать об основных признаках классических вариантов гомозиготных и гетерозиготных талассемий.
Online-консультации врачей
| Консультация гомеопата |
| Консультация сексолога |
| Консультация эндокринолога |
| Консультация оториноларинголога |
| Консультация анестезиолога |
| Консультация диетолога-нутрициониста |
| Консультация неонатолога |
| Консультация вертебролога |
| Консультация семейного доктора |
| Консультация эндоскописта |
| Консультация ортопеда-травматолога |
| Консультация стоматолога |
| Консультация иммунолога |
| Консультация массажиста |
| Консультация радиолога (диагностика МРТ, КТ) |
Новости медицины
Футбольные фанаты находятся в смертельной опасности,
31.01.2020
«Умная перчатка» возвращает силу хвата жертвам травм и инсультов,
28.01.2020
Назван легкий способ укрепить здоровье,
20.01.2020
Топ-5 салонов массажа в Киеве по версии Покупон,
15.01.2020
Новости здравоохранения
Глава ВОЗ объявил пандемию COVID-19,
12.03.2020
Коронавирус атаковал уже более 100 стран, заразились почти 120 000 человек,
11.03.2020
Коронавирус атаковал 79 стран, число жертв приближается к 3200 человек,
04.03.2020
Новый коронавирус атаковал 48 стран мира, число жертв растет,
27.02.2020
Прогноз течения болезни
Пациенты с умеренной и тяжелой формой имеют хорошие шансы на выживание, их главная задача – придерживаться правильной программы лечения (трансфузионная терапия и профилактика уровней железа с помощью комплексообразующих агентов). Сердечно-сосудистые заболевания из-за избытка железа — основная причина смерти. Трансплантация костного мозга способна излечить больного. Пациентам с талассемией, возможно, потребуется хирургическое вмешательство, чтобы исправить скелетные деформации.
Репортаж про ученных, которые научились бороться с данным заболеванием
https://youtube.com/watch?v=wGnaKqLJ2ao
Больным требуется постоянная диагностика общего анализа крови и уровней железа
Важно проверять печень (из-за развития гемохроматоза) и сердце, сдавать анализы на вирусные инфекции
Клинические проявления бета-талассемии
Лишь в редких случаях бессимптомного течения имеющиеся признаки не позволяют в раннем возрасте определить у ребенка наличие этого заболевания. Как правило, когда речь идет о бета-талассемии, симптомы во многом зависят от формы течения болезни. Таким образом, в некоторых случаях проявления будут крайне слабыми, а в других очень тяжелыми. Достаточно часто диагностируется большая бета-талассемия. Эта форма патологии получила название лихорадки Кули, протекает крайне тяжело и сопровождается появлением серьезных нарушений. В большинстве случаев симптомы очевидны уже в первые дни жизни малыша. Своего апогея они достигают примерно к 6 месяцам. К характерным признакам развития бета-талассемии относятся:
- снижение показателей гемоглобина до 60-20г/л;
- тяжелая анемия;
- пожелтение кожи;
- сильная бледность и даже синюшность;
- нарушение формирования костной ткани;
- изменения формы лица и черепа;
- замедление развития;
- увеличение печени и селезенки;
- частые инфекционные и воспалительные заболевания;
- проявления вторичного геморрагического синдрома.
У большинства детей, которые не погибают в первые полгода жизни, на фоне развития большой бета-талассемии наблюдается появление тяжелых нарушений работы эндокринной системы. Может проявиться карликовость и задержка полового созревания. Прогноз при этой форме заболевания крайне неблагоприятный и даже при частых переливаниях крови и выполнении других медицинских процедур продолжительность жизни больных редко превышает 20 лет. Примерно 30% детей умирают в первый год жизни. Большая часть больных доживает при правильном лечении до 8 лет.
Кроме того, достаточно распространенной является малая бета-талассемия. В этом случае выявляется мутация исключительно в 11-й паре хромосом. В результате этого у больных наблюдается лишь легкая анемия, причем уровень гемоглобина лишь незначительно понижен. К характерным клиническим проявлениям малой формы бета-талассемии относятся:
- расширение эритроцидного ростка в костном мозге;
- микроцитоз;
- низкое содержание гемоглобина в эритроцитах;
- анизопойкилоцидоз;
- незначительное увеличение селезенки.
Нередко эта форма болезни протекает без каких-либо серьезных физиологических изменений. При малой талассемии у детей нет проявлений, отражающихся на общем состоянии здоровья. Возможна легкая анемия, так как количество эритроцитов незначительно ниже нормы. Кроме того, в результатах анализов крови может обнаруживаться повышенный уровень железа и некоторые другие вещества, указывающие на наличие проблемы. В этом случае дети нормально развиваются, но при этом диагностика крайне важна, так как в дальнейшем уже взрослым людям необходимо планировать семью и рождение детей, чтобы их потомки были здоровыми.
Промежуточная бета-талассемия подтверждается, когда у больного в одном гене имеется слабая мутация, а в другом — тяжелая. В этом случае признаки болезни достаточно размыты, и нельзя на 100% причислить ее ни к большой, ни к малой форме бета-талассемии. Как правило, у детей с первых дней жизни наблюдается пожелтение кожных покровов, но уровень гемоглобина находится на нижней границе нормы. При таком течении патологии гемотрансфузию нужно делать достаточно редко, так как характерных признаков анемии не наблюдается.
https://youtube.com/watch?v=_sMBYX4rhHQ
В некоторых случаях может выявляться сильно повышенный уровень железа, что является показанием для удаления селезенки. Больному требуется постоянное наблюдение со стороны врача, так как вполне возможен переход бета-талассемии к более тяжелому течению
При таком варианте патологии больным очень важно придерживаться здорового образа жизни, так как вредные привычки могут спровоцировать ухудшение состояния
Разновидности бета-талассемии
Клиническая картина заболевания может быть разной, в зависимости от этого бета-талассемию подразделяют на несколько групп. Не все знакомы с таким понятием, как талассемия, что это заболевание зависит от многих генетических факторов, также известно далеко не всем.
Выделяют несколько состояний генов, которые контролируют выработку бета-цепей:
- Нормальный ген. Именно в таком состоянии он находится у всех здоровых людей.
- Практически разрушенный мутацией ген. Бета-цепь совсем не синтезируется.
- Частично поврежденный ген может только отчасти выполнять свою работу, поэтому синтез цепей идет, но в недостаточном количестве.
Учитывая все это, выделяют следующие виды талассемии:
- Талассемия минор. Легкая форма заболевания, формируется под воздействием всего одного поврежденного гена. По внешним показателям человек совершенно здоров. Только в анализах крови диагностируется небольшая анемия и маленький размер эритроцитов.