Синдром альпорта: развитие, признаки, диагностика, как лечить, прогноз
Содержание:
- Морфология
- Как диагностируют синдром Альпорта у детей?
- Лечение синдрома Альпорта
- Диагностика синдрома Альпорта
- Особенности клинической картины и лечения синдрома Альпорта у детей
- Особенности развития болезни у девушек и влияние на репродуктивную функцию
- Pathophysiology[edit]
- Лечение
- Симптомы
- Клиническая картина
- Особенности состояния
- Диагностика
Морфология
При световой микроскопии почечная ткань, полученная на ранних стадиях СА, выглядит нормальной. Фокальное и сегментарное утолщения капиллярных стенок клубочков, лучше выявляемые при окрашивании серебром, становятся видимыми при прогрессировании болезни. Они сочетаются с неспецифическими тубулярными поражениями и интерстицильным фиброзом. Стандартная иммунофлюоресценция, как правило, дает отрицательный результат. Однако могут обнаруживаться слабые и/или фокальные отложения иммуноглобулинов классов G и М и/или фракция комплемента СЗ. Основные повреждения выявляются ультраструктурным методом. Они характеризуются утолщением ГБМ (до 800—1200нм) с расщеплением и фрагментацией lamina densa на несколько волокон, образующих сеть наподобие корзины. Изменения ГБМ могут быть фрагментарными (неоднородными), чередующимися с участками нормальной или уменьшенной толщины. В целом, наиболее выраженной чертой у детей является неравномерное чередование очень толстых и очень тонких участков ГБМ. Диффузное истончение ГБМ обнаруживается примерно у 20% пациентов с СА.
На генетическом уровне СА — гетерогенное заболевание: мутации COL4A5 на Х-хромосоме связаны с Х-сцепленным СА, в то время как мутации COL4A3 или COL4A4 на 2-й хромосоме связаны с аутосомными формами болезни.
Х-сцепленный синдром Альпорта.
Как диагностируют синдром Альпорта у детей?
Предложены следующие критерии:
- Наличие в каждой семье не менее двух больных нефропатией,
- Гематурия как ведущий симптом нефропатии у пробанда,
- Наличие тугоухости хотя бы у одного из членов семьи,
- Развитие ХПН у одного родственника и более.
При диагностике разнообразных наследственных и врожденных заболеваний большое место принадлежит комплексному подходу к обследованию и прежде всего обращение внимания на данные, получаемые при составлении родословной ребенка. Диагноз синдрома Альпорта считается правомочным в случаях обнаружения у больного 3 из 4 типичных признаков
: наличие в семье гематурии и хронической почечной недостаточности, присутствие у больного нейросенсорной тугоухости, патологии зрения, обнаружение при электронно-микроскопической характеристике биоптата признаков расщепления гломерулярной базальной мембраны с изменением ее толщины и неравномерностью контуров .
Клинико-генетические методы исследования синдрома Альпорта
Прежде чем начнется лечение, проводится обследование больного, которое должно включать клинико-генетические методы исследования, направленное изучение анамнеза заболевания, общий осмотр больного с учетом диагностически значимых критериев.
- В стадии компенсации уловить патологию можно лишь ориентируясь на такие синдромы, как наличие наследственной отягощенности, гипотонии, множественных стигм дизэмбриогенеза, изменения мочевого синдрома.
- В стадии декомпенсации возможно появление эстраренальных симптомов, таких как выраженная интоксикация, астенизация, отставание в физическом развитии, анемизация, проявляющиеся и усиливающиеся с постепенным снижением почечных функций. У большинства больных при снижении почечных функций наблюдается: снижение функции ацидо- и аминогенеза, у 50% больных отмечают значительное снижение секреторной функции почек, ограничение пределов колебания оптической плотности мочи, нарушение ритма фильтрации, а затем и снижение клубочковой фильтрации.
- Стадия хронической почечной недостаточности диагностируется при наличии у больных в течение 3-6 мес. и более повышенного уровня мочевины в сыворотке крови (более 0,35 г/л), снижение клубочковой фильтрации до 25% от нормы.
Дифференциальная диагностика синдрома Альпорта
Ее приходится проводить с гематурической формой приобретенного гломерулонефрита. Приобретенный гломерулонефрит имеет чаще острое начало, период 2 — 3 недели после перенесенной инфекции, экстраренальные признаки, в том числе гипертензию с первых дней (при синдроме Альпорта, напротив, гипотония), снижение клубочковой фильтрации в начале заболевания, отсутствие нарушения парциальных канальцевых функций, тогда как при наследственном они присутствуют. Приобретенный гломерулонефрит протекает с более выраженной гематурией и протеинурией, с увеличенной СОЭ. Диагностическое значение имеют типичные изменения гломерулярной базальной мембраны, свойственные синдрому. Лечение должно быть начато оперативно.
Дифференциальная диагностика от дисметаболической нефропатии проводится с хронической почечной недостаточностью, в семье клинически выявляются неоднотипные болезни почек, а может быть спектр нефропатии от пиелонефрита до мочекаменной болезни. Часто присутствуют жалобы на боли в животе и периодически при мочеиспускании, в осадке мочи – оксалаты.
При подозрении на заболевание, больного необходимо направить для уточнения диагноза в специализированное нефрологическое отделение.

Лечение синдрома Альпорта
В режиме предусматривается ограничение от больших физических нагрузок, пребывание на свежем воздухе. Диета полноценная, с достаточным содержанием полноценных белков, жиров и углеводов с учетом функции почек. Большое значение имеет выявление и санация хронических очагов инфекции. Из лекарственных средств используются АТФ, кокарбоксилаза, пиридоксин (до 50 мг/сут.), карнитина хлорид. Курсы проводят 2-3 раза в год. При гематурии назначается фитотерапия — крапива двудомная, сок черноплодной рябины, тысячелистник.
В зарубежной и отечественной литературе имеются сообщения о лечении преднизолоном и использовании цитостатиков. Однако об эффекте судить трудно.
При хронической почечной недостаточности применяются гемодиализ и трансплантация почек.
Методов специфической (эффективной патогенетической) терапии наследственного нефрита не существует. Все лечебные мероприятия направлены на предупреждение и замедление снижения почечных функций.
Диета должна быть сбалансированной и высококалорийной, с учетом функционального состояния почек. При отсутствии нарушений функционального состояния в питании ребенка должно быть достаточное содержание белков, жиров и углеводов. При наличии признаков почечных дисфункции количество белка, углеводов кальция и фосфора следует ограничивать, что отдаляет сроки развития хронической почечной недостаточности.
Физические нагрузки должны быть ограничены, детям рекомендуется отказ от занятий спортом.
Следует избегать контактов с инфекционными больными, снижать риск развития острых респираторных заболеваний. Необходима санация очагов хронической инфекции. Профилактические прививки детям с наследственным нефритом не проводятся, вакцинация возможна только по эпидемиологическим показаниям.
Гормональная и иммуносупрессивная терапия при наследственном нефрите неэффективна. Есть указания на некоторый положительный эффект (снижение уровня протеинурии и замедление прогрессирования заболевания) при длительном многолетнем применении циклоспорина А и ингибиторов АПФ.
В терапии больных используют препараты, улучшающие обмен:
- пиридоксин — по 2-3 мг/кг/сут в 3 приема в течение 4 недель;
- кокарбоксилаза — по 50 мг внутримышечно через день, всего 10-15 инъекций;
- АТФ — по 1 мл внутримышечно через день, 10-15 инъекций;
- витамин А — по 1000 ЕД/год/сут в 1 прием в течение 2 недель;
- витамин Е — по 1 мг/кг/сут в 1 прием в течение 2 недель.
Подобная терапия способствует улучшению общего состояния больных, снижению тубулярных дисфункций и проводится курсами 3 раза в год.
В качестве иммуномодулятора может быть использован левамизол — по 2 мг/кг/сут 2-3 раза в неделю с перерывами между приемами в 3-4 дня.
Поданным исследований положительный эффект на выраженность гематурии и нарушений функции почек оказывает гипербарическая оксигенация.
Наиболее эффективным методом лечения наследственного нефрита является своевременная трансплантация почки. При этом не отмечается рецидива заболевания в трансплантате, в небольшом проценте случав (около 5%) возможно развитие нефрита в трансплантированной почке, связанного с антигенами к гломерулярной базальной мембране.
Диагностика синдрома Альпорта
Предложены следующие критерии:
- наличие в каждой семье не менее двух больных нефропатией;
- гематурия как ведущий симптом нефропатии у пробанда;
- наличие тугоухости хотя бы у одного из членов семьи;
- развитие хронической почечной недостаточности у одного родственника и более.
При диагностике разнообразных наследственных и врожденных заболеваний большое место принадлежит комплексному подходу к обследованию и прежде всего обращение внимания на данные, получаемые при составлении родословной ребенка. Диагноз синдрома Альпорта считается правомочным в случаях обнаружения у больного 3 из 4 типичных признаков: наличие в семье гематурии и хронической почечной недостаточности, присутствие у больного нейросенсорной тугоухости, патологии зрения, обнаружение при электронно-микроскопической характеристике биоптата признаков расщепления гломерулярной базальной мембраны с изменением ее толщины и неравномерностью контуров.
Обследование больного должно включать клинико-генетические методы исследования; направленное изучение анамнеза заболевания; общий осмотр больного с учетом диагностически значимых критериев. В стадии компенсации уловить патологию можно лишь ориентируясь на такие синдромы, как наличие наследственной отягощенности, гипотонии, множественных стигм дизэмбриогенеза, изменения мочевого синдрома. В стадии декомпенсации возможно появление эстраренальных симптомов, таких как выраженная интоксикация, астенизация, отставание в физическом развитии, анемизация, проявляющиеся и усиливающиеся с постепенным снижением почечных функций. У большинства больных при снижении почечных функций наблюдается: снижение функции ацидо- и аминогенеза; у 50% больных отмечают значительное снижение секреторной функции почек; ограничение пределов колебания оптической плотности мочи; нарушение ритма фильтрации, а затем и снижение клубочковой фильтрации. Стадия хронической почечной недостаточности диагностируется при наличии у больных в течение 3-6 мес и более повышенного уровня мочевины в сыворотке крови (более 0,35 г/л), снижение клубочковой фильтрации до 25% от нормы.
Дифференциальную диагностику наследственного нефрита приходится проводить прежде всего с гематурической формой приобретенного гломерулонефрита. Приобретенный гломерулонефрит имеет чаще острое начало, период 2-3 нед после перенесенной инфекции, экстраренальные признаки, в том числе гипертензию с первых дней (при наследственном нефрите, напротив, гипотония), снижение клубочковой фильтрации в начале заболевания, отсутствие нарушения парциальных канальцевых функций, тогда как при наследственном они присутствуют. Приобретенный гломерулонефрит протекает с более выраженной гематурией и протеинурией, с увеличенной СОЭ. Диагностическое значение имеют типичные изменения гломерулярной базальной мембраны, свойственные наследственному нефриту.
Дифференциальная диагностика от дисметаболической нефропатии проводится с хронической почечной недостаточностью, в семье клинически выявляются неоднотипные болезни почек, а может быть спектр нефропатии от пиелонефрита до мочекаменной болезни. У детей часто присутствуют жалобы на боли в животе и периодически при мочеиспускании, в осадке мочи — оксалаты.
При подозрении на наследственный нефрит больного необходимо направить для уточнения диагноза в специализированное нефрологическое отделение.
Особенности клинической картины и лечения синдрома Альпорта у детей
Заподозрить наличие патологии у малыша можно сразу же после его появления на свет. Дети имеют характерные изменения и деформации лицевого черепа в виде расщепления верхней губы (заячья губа) и воронкообразного твёрдого нёба (волчья пасть), увеличенные размеры головы, а также типичное выражение лица с блуждающим взглядом. Другие мутации могут проявляться в форме нарушения слуха и зрения. Такой ребёнок не реагирует даже на громкие звуки и резкие движения окружающей среды, что обнаруживается ещё в родильном доме.
Для устранения внешних дефектов маленьким пациентам проводится пластическая операция. Мне довелось участвовать в ушивании верхней губы ребёнку с такой проблемой. После хирургического вмешательства остаётся небольшой шрам, который со временем бледнеет и становится практически незаметным. Операции проводятся в течение нескольких недель после рождения ребёнка, что снижает риск возникновения вторичных осложнений.
Для профилактики повреждений почечной ткани маленьким пациентам назначается специальная диета. Новорождённые и младенцы находятся на грудном вскармливании или получают максимально адаптированную к составу женского молока смесь. Это позволяет устранить риск развития иммунного дефицита. Основные средства для терапии симптоматических признаков заболевания вводятся на 5–7 году жизни (раньше — по потребности) и практически не отличаются от лечения, которое получает взрослый человек. Для адаптации слепых и глухих детей в обществе используют слуховые аппараты, корректирующие операции. Первые несколько лет ребёнок должен посещать специальные школы или дополнительно заниматься со специалистом, чтобы нагнать сверстников.
Особенности развития болезни у девушек и влияние на репродуктивную функцию
У пациентов женского пола не всегда встречаются явные признаки синдрома Альпорта. Многие девушки узнают о носительстве подобной патологии только после рождения больного малыша. В более тяжёлых случаях заболевание активизируется в период полового созревания под воздействием гормональной перестройки организма (во время менструации) или после первого сексуального опыта.
В моей практике встречалась пациентка, у которой синдром Альпорта появлялся через каждые 3 поколения. У данной больной патология длительное время пребывала в спящем состоянии, но активизировалась после перенесённого стресса. Женщина отметила, что от этой формы недуга страдали также её прабабка и родная сестра. Доктора рассчитали, что вероятность появления подобной болезни у последующих поколений крайне велика.
Пациентки с тяжёлой формой синдрома Альпорта испытывают определённые трудности при зачатии и вынашивании ребёнка. Это может быть связано как с аномалиями строения репродуктивных органов, так и с нарушением работы эндокринной системы. В период роста и развития плода в материнском организме вырабатывается в 2 раза больше жидкости, в результате чего существенно возрастает нагрузка на почки. Из-за нефритических изменений воспалительного характера организм не справляется с увеличением объёма циркулирующей крови, а у женщины возникают выкидыши на различных сроках.
Какие осложнения могут возникнуть у пациенток с синдромом Альпорта при вынашивании беременности:
- Развитие преэклампсии — состояния, сопровождающегося выделением с мочой белка и резким повышением артериального давления до критических цифр. Эта патология представляет собой серьёзную угрозу для жизни не только плода, но и матери. Всех пациенток с подобным диагнозом госпитализируют в стационар и подбирают им оптимальную методику родоразрешения.
-
Выкидыши или невынашивание беременности. Из-за проблем с почками у матери ребёнок появляется на свет намного раньше заявленного срока. При массе более 500 грамм таких детей выхаживают в реанимации, но из-за внутриутробного недоразвития они немного отличаются в физическом и психическом плане от своих сверстников.
- Создание условий для передачи инфекции через плаценту. У пациенток с синдромом Альпорта значительно снижается функция иммунной системы, что усиливается во время беременности. Это приводит к увеличению различных внутриутробных патологий, которые возникают из-за присоединения инородной микрофлоры. Лечение такого малыша происходит в реанимации новорождённых.
- Преждевременная отслойка нормально расположенной плаценты (детского места). При синдроме Альпорта из-за поражения почек часто развиваются явления артериальной гипертензии (рост давления). Усиленный ток крови может способствовать повреждению сосудистых образований плаценты и её отрыву. Пациентки с таким осложнением должны быть экстренно госпитализированы в реанимационное отделение для проведения оперативного вмешательства.
Наследственные заболевания почек — это группа опасных патологий которые существенно снижают не только качество жизни человека, но и её продолжительность. В настоящее время особенно актуальной является проблема ранней диагностики синдрома Альпорта. Почти все пациенты живут в неведении в течение многих лет, пока какой-либо резкий фактор не спровоцирует развитие клинических симптомов заболевания и его осложнений
Именно поэтому так важно постоянно следить за своим здоровьем, отмечая малейшие изменения в самочувствии, а также регулярно обращаться к доктору для контроля анализов
Pathophysiology[edit]
Genetics
Alport syndrome is caused by mutations in COL4A3, COL4A4, and COL4A5, three of six human genes involved in basement membrane (type IV) collagen biosynthesis. Mutations in any of these genes prevent the proper production or assembly of the specialised type IV collagen ‘345’ network which is an important structural component of basement membranes in the kidney, inner ear, and eye. It is also found in other locations, including the alveoli of the lungs. Basement membranes are thin, sheet-like structures that separate and support cells in many tissues. Type IV collagen ‘112’ type is found in both vertebrates and invertebrates, and is the major isoform in most human basement membranes. When mutations prevent the formation of 345 type IV collagen network in the glomerulus, the 112 network, which is formed in fetal development but usually replaced by 345, persists into adult life.[citation needed]
Inheritance patterns
Alport syndrome can have different inheritance patterns depending on which specific mutation is present.
- In most people with Alport syndrome (about 85%), the condition is inherited in an X-linked pattern, due to mutations in the COL4A5 gene. A condition is considered X-linked if the gene involved in the disorder is located on the X chromosome. In males, who have only one X chromosome, one altered copy of the COL4A5 gene is sufficient to cause severe Alport syndrome, explaining why most affected males eventually develop kidney failure. In females, who have two X chromosomes, a mutation in one copy of the COL4A5 gene usually results in blood in the urine, but most affected females do not develop kidney failure.
- Alport syndrome can also be inherited in an autosomal recessive pattern if both copies of the COL4A3 or COL4A4 gene, located on chromosome 2, have been mutated. Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.[citation needed]
- Past descriptions of an autosomal dominant form are now usually categorized as other conditions. Notably, conditions associated with giant platelets and associated with mutations of MYH9 are no longer considered to be Alport variants. However apparent autosomal dominant transmission of disease associated with mutations in COL4A3 and COL4A4 does occur.
Clinical utility gene card for: Alport syndrome.
Лечение
Тактика лечения: специфического лечения нет, симптоматическое лечение.
Цели лечения:
— активизация психического развития;
— пополнение пассивного и активного словарного запаса;
— коррекция поведения;
— повышение эмоционального тонуса, настроения ребенка;
— обучение навыкам самообслуживания;
— социальная адаптация.
Немедикаментозное лечение:
— индивидуальные занятия с логопедом;
— занятия с психологом;
— кондуктивная педагогика;
— малокалорийная диета;
— ЛФК;
— физиолечение.
Медикаментозное лечение
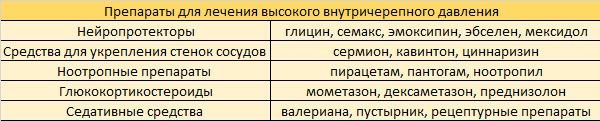
Широко используют в последнее время препараты ноотропного ряда — нейропротекторы, с целью улучшения обменных процессов в головном мозгу. Большинство ноотропных препаратов в связи с их психостимулирующим действием назначают в первую половину дня. Продолжительность курсов лечения ноотропами составляет от одного до двух-трех месяцев, церебролизин, ампулы 1 мл в/м, пирацетам, ампулы 5 мл 20%, гинкго-билоба (танакан), таблетки 40 мг, пиритинол гидрохлорид (энцефабол), драже 100 мг, суспензия — 5 мл содержат 80,5 мг пиритинола (соотв.100 мг пиритинола гидрохлорида).
Энцефабол — минимум противопоказаний, разрешен к применению с первого года жизни. Дозирование суспензии (с содержанием в 1 мл 20 мг энцефабола) детям 3-5 лет суточная доза 200-300 мг (12-15 мг массы тела) назначают в 2 приема — утром (после завтрака) и днем (после дневного сна и полдника). Продолжительность курса 6-12 недель (целесообразен длительный прием, при котором повышается работоспособность и способность к обучению, улучшаются высшие психические функции): актовегин, ампулы 2 мл 80 мг, драже-форте 200 мг активного вещества.
Нейрометаболический препарат, содержащий исключительно физиологические компоненты. Детям назначается в драже-форте, прием до еды по ½ -1 драже 2-3 раза в день (в зависимости от возраста и выраженности симптомов заболевания) до 17 часов. Продолжительность терапии 1-2 месяца. Инстенон, таблетки (1 таблетка содержит 50 мг этамивана, 20 мг гексобендина, 60 мг этофиллина). Многокомпонентный нейрометаболический препарат. Суточная доза составляет 1,5-2 таблетки, назначается в 2 приема (утром и днем) после еды. Для исключения побочных эффектов рекомендуется постепенное наращивание дозы в течение 5-8 дней. Продолжительность лечения 4-6 недель.
Ангиопротекторы с целью улучшения мозгового кровообращения: винпоцетин, циннаризин.
Седативная терапия по показаниям: ноофен, ново-пассит.
Корректоры поведения: сонапакс, хлорпротиксен.
Витамины группы В: В1, В6, В12, нейромультивит, неуробекс, фолиевая кислота, аевит.
Профилактические мероприятия:
— профилактика травматизма;
— профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение: регулярные занятия с логопедом, дефектологом, психологом, социальная адаптация ребенка, оформление в специализированный детский сад, прохождение медико-педагогической комиссии для решения вопроса об обучении ребенка.
Основные медикаменты:
1. Актовегин, ампулы 2 мл 80 мг
2. Винпоцетин, таблетки 5 мг
3. Пирацетам, ампулы 5 мл 20%
4. Пирацетам, таблетки 0,2 и 0,4
5. Пиридоксин гидрохлорид, ампулы, 1 мл 5%
6. Танакан, таблетки 40 мг
7. Тиамин хлорид, ампулы 5% 1 мл
8. Фолиевая кислота, таблетки 0,001
9. Церебролизин, ампулы 1 мл
10. Цианокобаламин, ампулы 1 мл 200 мкг и 500 мкг
Дополнительные медикаменты:
1. Аевит, капсулы
2. Глицин, таблетки 0,1
3. Гопантеновая кислота (пантокальцин), таблетки 0,25
4. Инстенон, таблетки
5. Нейромультивит, таблетки
6. Неуробекс, таблетки
7. Ноофен,таблетки 0,25
8. Пиритинол, драже 0,1, суспензия
9. Сонапакс, таблетки 10 мг
10. Хлорпритиксен 15 мг
11. Циннаризин, таблетки 25 мг
Индикаторы эффективности лечения:
— улучшение внимания, памяти, работоспособности;
— пополнение пассивного и активного запаса слов;
— повышение эмоционального и психического тонуса.
Симптомы
В три года и раньше у ребенка появляются первые симптомы, выраженные изолированным мочевым синдромом, то есть гематурией (изменением количества или качества мочи, её осадка). Чаще всего наследственный нефрит у детей выявляется во время обследования перед поступлением в детские сады или при ОРВИ, то есть случайно. Семейный нефрит отличается от приобретенного отсутствием временного промежутка между началом действия раздражителя и ответной реакции.
Дети не чувствуют дискомфорта на начальной стадии хвори. Главным показателем болезни является измененный цвет мочи, смена пигмента всегда наблюдается на первых этапах. Усиленная пигментация в красные тона провоцируется прививками, фитнес нагрузкой, заболеваниями гортани, легких. На начальных этапах превышения выделения белка с мочой непостоянны, всё же протеинурия возрастает с течением болезни. Возможно наличие лейкоцитурии — признак деструктивных изменений в организме.
Следующим этапом идет ухудшение самочувствия пациента: отравление организма, слабость, пониженное давление, развитие глухоты, возможны проблемы с глазами. Понижение чувствительности слухового аппарата на начальных этапах выявляется только при помощи исследования слухового аппарата. Самым распространенным возрастом начала проявления глухоты является промежуток с шести до десяти лет. Тугоухость может являться первым симптомом заболевания, может проявиться даже раньше мочевого синдрома.
Зрение при наследственном нефрите портится с вероятностью двадцати процентов. Порча зрения выражается одним из вариантов, таких как: миопия, катаракта, изменение физического устройства хрусталика.
В подростковом возрасте явным симптомом является повышенное давление, физическая недоразвитость. Характерной стигмой болезни является смешение таких аномалий, как: деформация прикуса, неправильная форма ушей, аномальная кривизна мизинца руки, большой промежуток между первым и вторым пальцем стопы. Синдром Альпорта, приобретенный наследственным путем, характеризуется схожими отклонениями у родственников.
Если мы говорим про то, как пошагово происходят нарушения в структуре работы почек, то выделяется минимум три этапа. Первый: дестабилизация частичной функциональной работы почек. Второй: деструктивная работа как центральных, так и срединных частей почек. Третий: Ухудшение фильтрации клубочкового аппарата.
Подытоживая симптомы, приходим к выводу о том, что заболевание проходит «поступенчато», то есть поэтапно. Изначально мы имеем дело со скрытой бессимптомной болезнью, а потом она перетекает в открытое заболевание с явной симптоматикой.
Клиническая картина
Симптомы синдрома Альперса в связи с постоянным прогрессированием заболевания со временем становятся все отчетливее. Зачастую первые признаки можно увидеть уже у детей первых месяцев жизни. Отставание в психическом, умственном и физическом развитии выражается как:
задержка развития основных навыков. Ребенок не держит головку, не переворачивается, не берет в ручки предметы. Он вялый, наблюдается выраженный гипотонус мышц. Синдром Альперса у детей проявляется еще неумением разговаривать, эмоционально реагировать на раздражители;

Ребенок не держит головку, не переворачивается
- частые срыгивания и рвота;
- судорожный синдром. Судороги могут быть генерализованными (затрагивающими все тело) или локальными (на определенном участке организма), разной частоты и длительности;
- нарушения координации движений. Ребенок совершает резкие, размашистые движения, они лишены плавности и точности;
- эпилептические припадки. Часто они статусного характера, резистентные к терапии;
- тяжелые поражения печени. Очень быстро развивается цирроз, а впоследствии печеночная недостаточность. Этот симптом развивается в терминальной стадии синдрома.
Синдром Альперса у новорожденных проявляется скованностью суставов, деформацией грудной клетки, сниженной массой тела, внутриутробной задержкой роста и микроцефалией.
Симптомы постоянно прогрессируют.
Особенности состояния
Нефротический синдром – обширный симптомокомплекс, развитие которого обусловлено поражением почек и почечных структур различной тяжести. Состояние характеризуется появлением белка в моче, отечностью нижних конечностей и лица, дистрофией слизистых оболочек или кожных покровов. Существуют две основных формы нефротического синдрома:
- первичная, когда патология развивается изолированно (врожденный нефрит);
- вторичная, когда симптомокомплекс становится следствием провоцирующих факторов: инфекции, аутоиммунной патологии, воспалительного процесса, синдрома Альпорта.
Клиницисты выделяют генетическую форму нефротического синдрома, появление которой обусловлено наследственной предрасположенностью. По статистике, почти в 25% нефротический синдром значительно ухудшает функцию почек, осложняет течение основного урологического или нефрологического заболевания. Вторичный патологический синдром клинически обратим, но только при сохранении функции почек до 60–75%.
Диагностика
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, зрения, слуха, ожирение, расторможенность, врожденные пороки развития со стороны органов зрения, почек, опорно-двигательного аппарата; отягощенная наследственность, перинатальная патология.
Физикальное обследование
Синдром Альпорта — наследственная болезнь, характеризующаяся снижением остроты зрения (катаракта, пигментный ретинит, лентиконус, сферофакия), глухотой в связи с недоразвитием слуховых анализаторов, признаками нефрита (протеинурия, гематурия) и развитием в возрасте 16-35 лет почечной недостаточности. Наследуется по аутосомно-рецессивному типу, сцепленному с полом.
Синдром Лоренса-Муна-Бидля (фенотипические проявления) — основным симптомом заболевания является ожирение, пигментный ретинит, гипогенитализм, полидактилия, умственная отсталость. Полидактилия обнаруживается у 60-70% больных. Пигментная дегенерация сетчатки ведет к снижению зрения, обычно проявляющемуся к 6-7 годам и неуклонно прогрессирующее. Ожирение появляется еще раньше — на 1-2 году жизни и быстро прогрессирует. Среди других нарушений у больных отмечаются отставание в росте, глухота (5% случаев), врожденные пороки сердца, различные аномалии глаза (микрокорнеа, катаракта, микрофтальмия, макулярная дегенерация и другие). Часто имеются аномалии в строении черепа и лица (акроцефалия, широкое переносье, эпикант). Умственная отсталость от легкой до тяжелой.
Синдром Целвегера (церебро-гепато-ренальный синдром) — множественные признаки дисплазии (аномалии развития мозгового и лицевого черепа, глаз, ушей, половых органов), расстройство глотания, выраженная гипотония мышц, сухожильная арефлексия, нарушение психомоторного развития в сочетании с врожденной патологией печени и почек. Возможно в основе — расстройство аминокислотного обмена. Тип наследования — аутосомно-рецессивный.
Лабораторные исследования: общий анализ крови и мочи, кал на яйца глист, кариотип.
Инструментальные исследования
1. Электроэнцефалография (ЭЭГ): на ЭЭГ задержка формирования возрастной корковой ритмики, диффузные изменения электрогенеза головного мозга.
2. Электромиография (ЭМГ) — снижение амплитуды интерференционной кривой при произвольном мышечном сокращении.
3. Компьютерная томография головного мозга (КТ).
Показания для консультации специалистов:
— логопед;
— психолог;
— ЛОР сурдолог;
— окулист;
— кардиолог;
— генетик;
— педиатр;
— ортопед;
— эндокринолог.
Минимум обследования при направлении в стационар:
— общий анализ крови;
— общий анализ мочи;
— АЛТ;
— АСТ;
— кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
3. ЭЭГ.
4. Исследование слуха.
5. КТ головного мозга.
6. Логопед.
7. Психолог.
8. Окулист.
9. Кариотип.
10. Генетик.
11. Эндокринолог.
12. ЭКГ.
13. Кардиолог.
14. УЗИ органов брюшной полости.
Дополнительные диагностические мероприятия:
1. ИФА на токсоплазмоз.
2. ИФА на цитомегаловирус.
3. Анализ мочи на обменные нарушения.